Набухание полимеров
Набухание полимеров (swelling, Quellung, gonflement) — это увеличение объема (массы) полимеров в результате поглощения низкомолекулярной жидкости или ее паров. Неограниченным набуханием иногда называют процесс растворения полимера, однако правильнее применять термин «набухание» только в тех случаях, когда жидкость поглощается ограниченно, или к стадии, предшествующей растворению, поскольку, как правило, представление о набухании связывается с сохранением общей формы образца.
Количественные характеристики набухания:
- степень набухания
- скорость набухания.
Степень набухания (α) определяют отношением массы (объема) поглощенной полимером жидкости к массе (объему) исходного полимера (mo):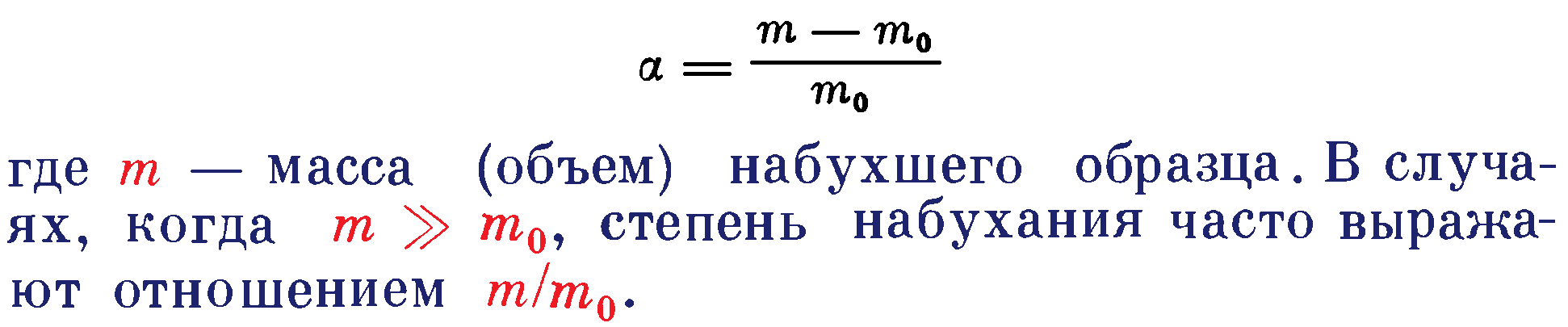
Скорость набухания dx/dτ (τ — продолжительность набухания) обычно имеет максимальное значение в начале процесса. При ограниченном набухании по мере приближения системы к состоянию равновесия dx/dτ—>0, а α—> αmах (αmах — равновесная, или максимальная, степень набухания).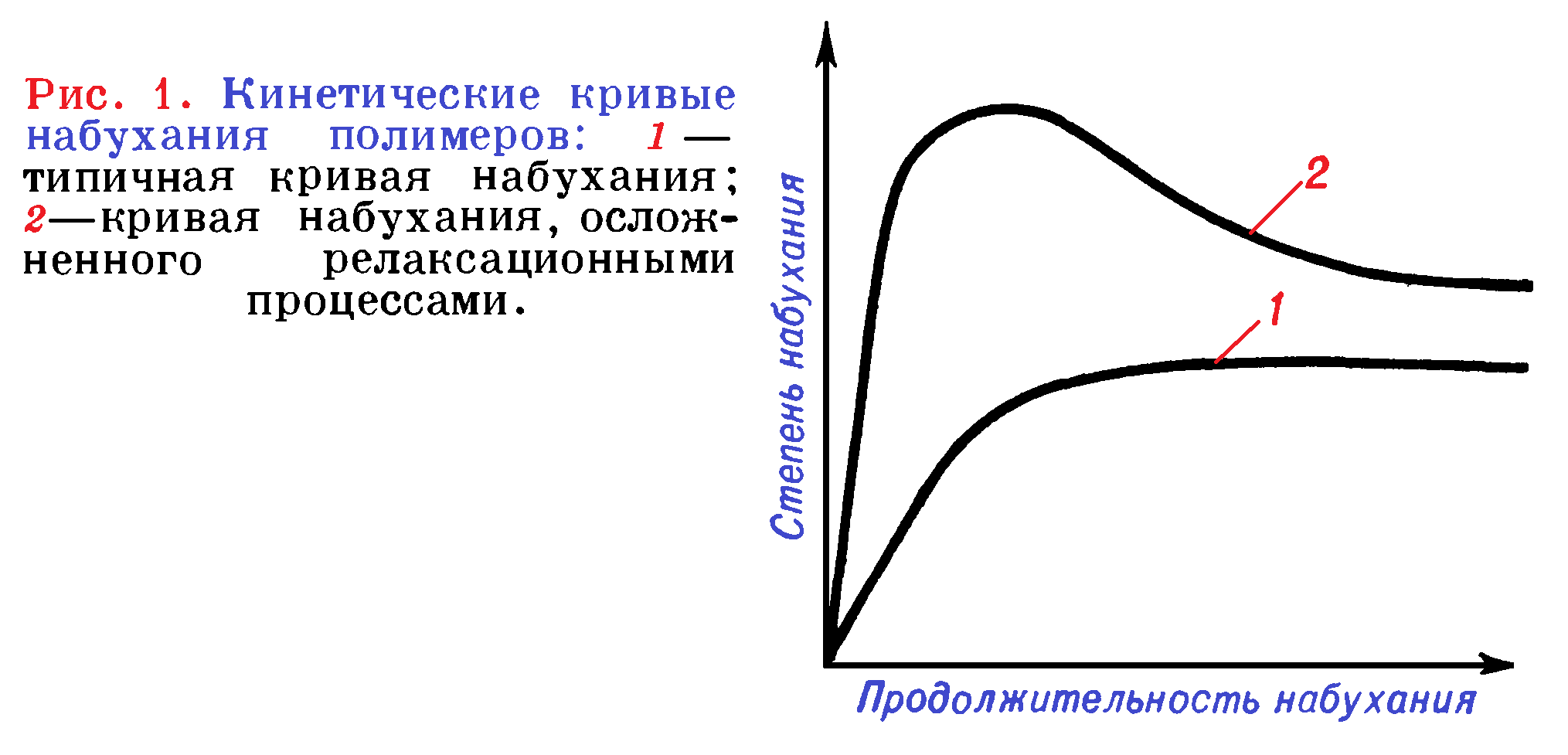
Кинетика набухания может быть описана различными уравнениями.
Для простейшего случая (кривая 1 на рисунке 1) широко используют дифференциальное уравнение 1 или его интегральную форму (уравнение 2):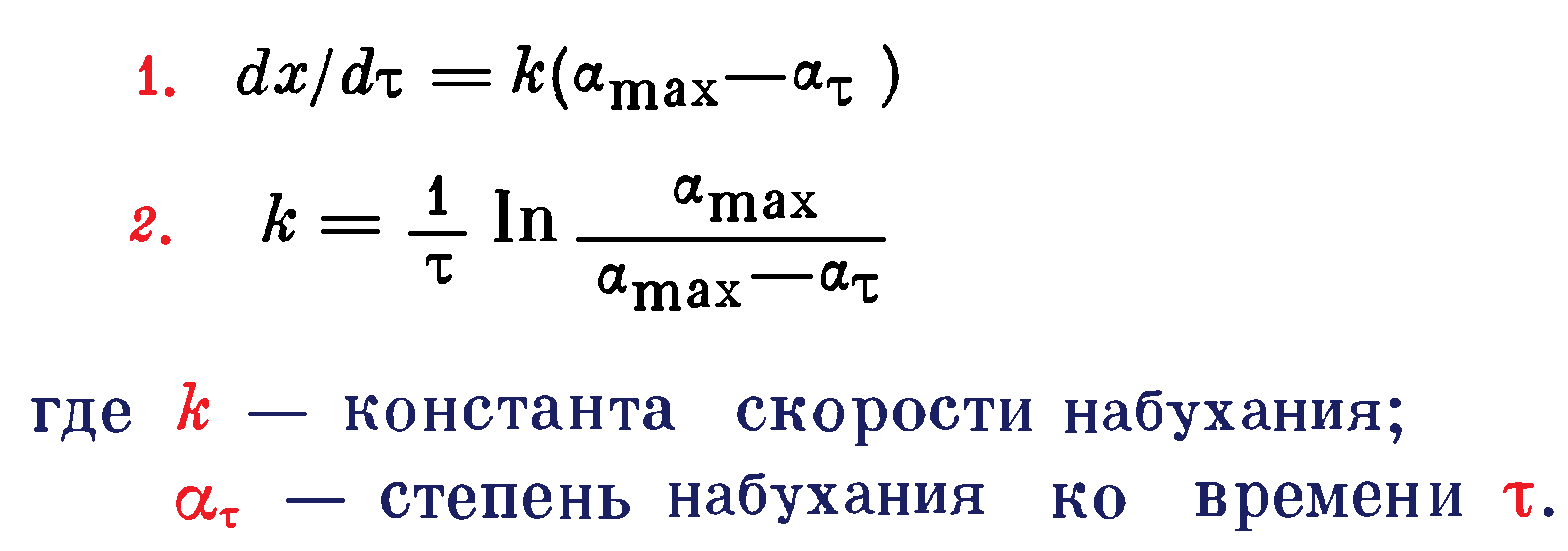
В некоторых случаях после достижения максимальной степени набухания (полимер «отдает» часть поглощенной жидкости в результате перестройки своей структуры и протекающих при этом релаксационных процессов (рисунок 1, кривая 2).
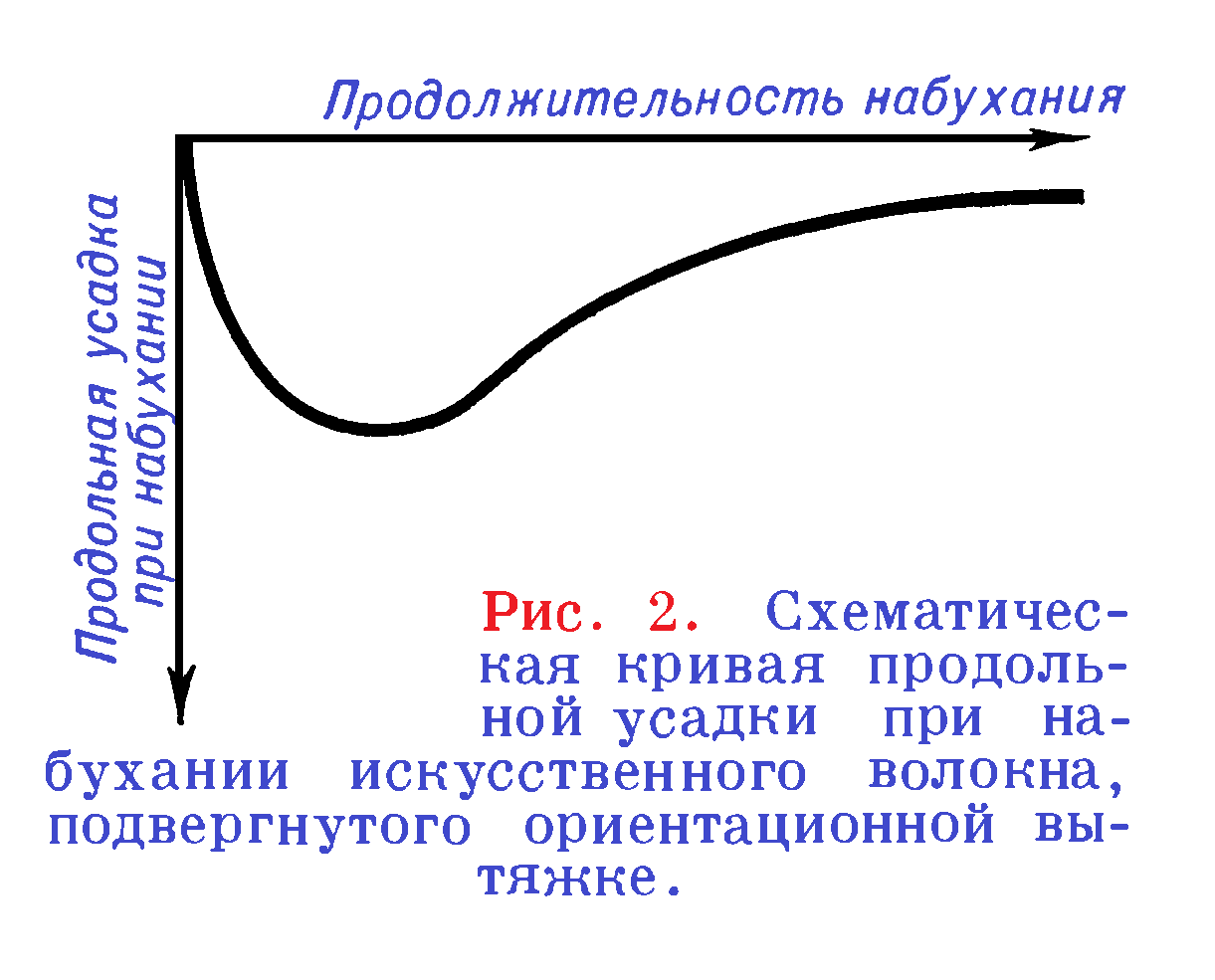 Изотропные полимерные материалы, у которых нет ориентации макромолекул в каком-либо направлении, сохраняют при набухании исходную форму. При набухании анизотропных полимерных материалов (пленок и особенно волокон) размеры образца в направлении, перпендикулярном оси ориентации, изменяются значительно больше, чем вдоль этой оси. В некоторых случаях наблюдается даже усадка материала вдоль оси. Количественно анизотропия набухания полимеров характеризуется отношением продольного удлинения (усадки) к изменению поперечных размеров. Это отношение может служить косвенным показателем степени ориентации полимерного материала. При набухании искусственных волокон, сохраняющих после формования внутренние напряжения, размеры вдоль оси волокна изменяются во времени по сложной кривой (рисунок 2). При этом степень набухания волокна на всех стадиях процесса больше 1 (здесь и далее за степень набухания принято отношение m/m0).
Изотропные полимерные материалы, у которых нет ориентации макромолекул в каком-либо направлении, сохраняют при набухании исходную форму. При набухании анизотропных полимерных материалов (пленок и особенно волокон) размеры образца в направлении, перпендикулярном оси ориентации, изменяются значительно больше, чем вдоль этой оси. В некоторых случаях наблюдается даже усадка материала вдоль оси. Количественно анизотропия набухания полимеров характеризуется отношением продольного удлинения (усадки) к изменению поперечных размеров. Это отношение может служить косвенным показателем степени ориентации полимерного материала. При набухании искусственных волокон, сохраняющих после формования внутренние напряжения, размеры вдоль оси волокна изменяются во времени по сложной кривой (рисунок 2). При этом степень набухания волокна на всех стадиях процесса больше 1 (здесь и далее за степень набухания принято отношение m/m0).
При набухании в условиях сохранения постоянного объема полимера внутри образца развиваются очень высокие давления. При поглощении полимером первых порций жидкости (до 3—5% по массе) давление набухания составляет несколько сот Мн/м2 (несколько тысяч кгс/см2), а при поглощении двух-четырехкратного количества оно уменьшается до 0,15—0,2 Мн/м2 (1,5—2 кгс/см2). Установлению равновесия отвечает нулевое давление набухания.
При взаимодействии полярных полимеров с полярными низкомолекулярными жидкостями поглощение первых порций жидкости сопровождается выделением довольно большого количества тепла; дальнейшее поглощение сопровождается незначительным тепловым эффектом. Набухание гибкоцепных неполярных полимеров в неполярных жидкостях не связано с существенными тепловыми эффектами, так как происходит в основном за счет изменения энтропии системы.
Набухание аморфных полимеров.
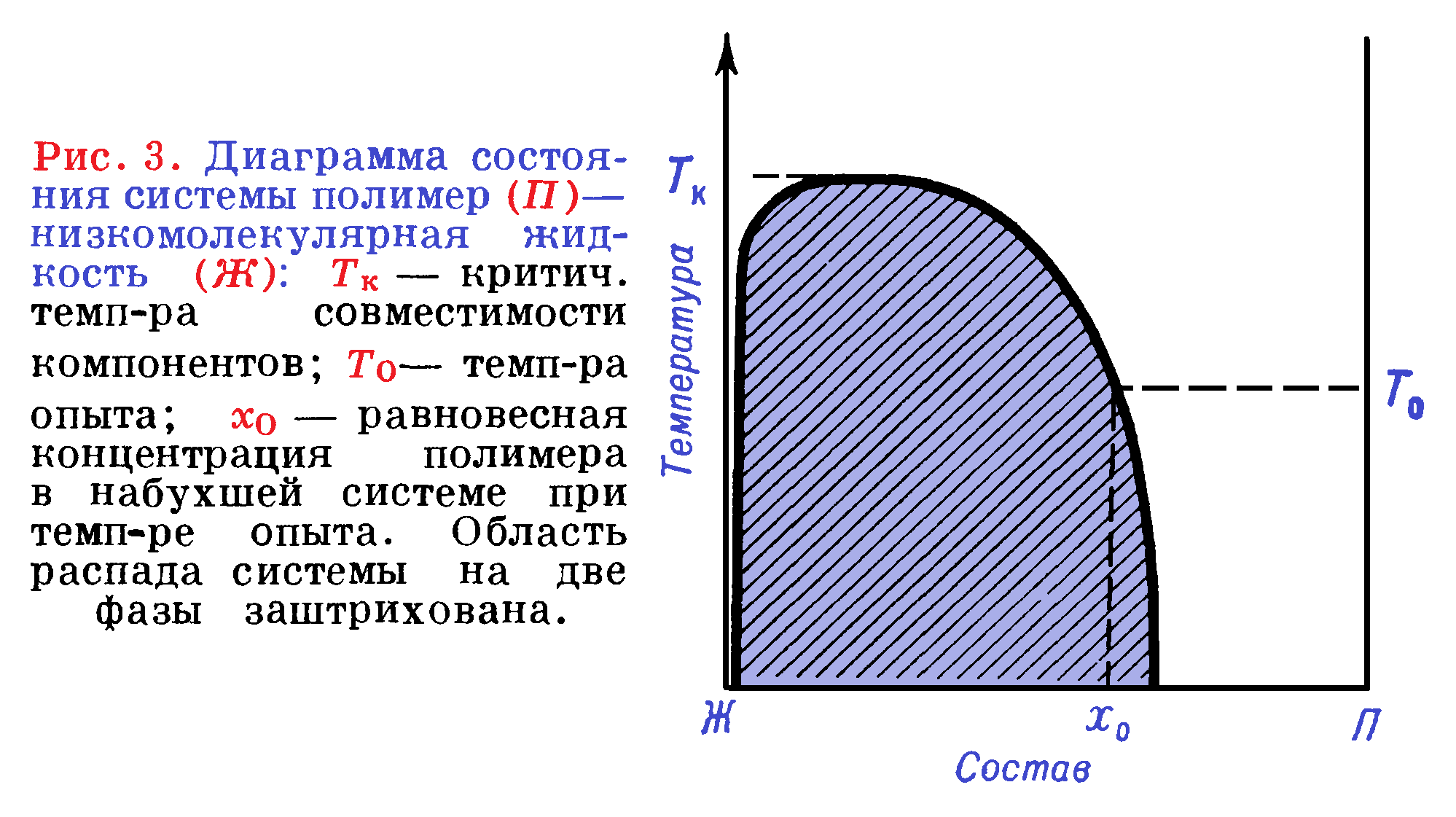 Теоретически степень набухания аморфного полимера, свободного от остаточных внутренних напряжений и с однородным молекулярно-массовым распределением по всему объему, определяется положением точки на диаграмме состояния системы полимер — низкомолекулярная жидкость (рисунок 3).
Теоретически степень набухания аморфного полимера, свободного от остаточных внутренних напряжений и с однородным молекулярно-массовым распределением по всему объему, определяется положением точки на диаграмме состояния системы полимер — низкомолекулярная жидкость (рисунок 3).
Если через х0 обозначена доля полимера (по массе) в системе при температуре T0, то степень набухания по массе (в %) составит — (1/ х0)·100. Выше критической температуры смешения понятие о равновесной степени набухания не имеет смысла так как полимер и низкомолекулярная жидкость совместимы в любых соотношениях.
Состояние равновесия при набухании аморфного полимера термодинамически отвечает равенству химических потенциалов низкомолекулярной жидкости в чистом виде и в набухшем полимере. Для чистой низкомолекулярной жидкости относительное давление паров над набухшим полимером в положении равновесия равно 1. Снижение этого давления в результате введения в жидкость каких-либо растворимых веществ должно приводить, в принципе, к уменьшению степени набухании полимера.
Это явление осложняется, однако, если вводимый в жидкость компонент активно взаимодействует с полимером (например, является растворителем для полимера).
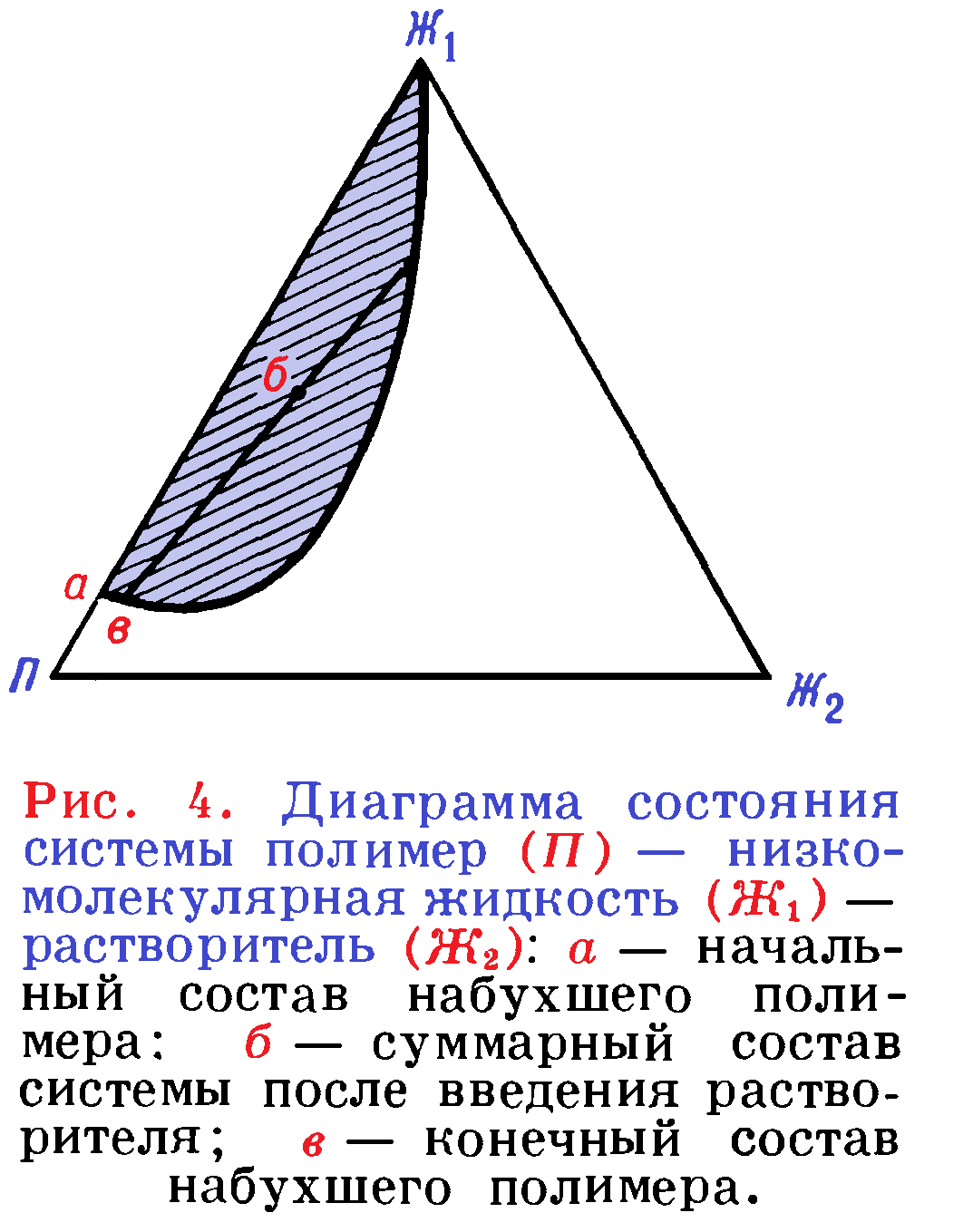 Схематически изменение степени набухания при введении нового компонента можно передать диаграммой состояния трехкомпонентной системы (рисунок 4). Пусть чистая низкомолекулярная жидкость Ж1 вызывает набухание полимера П до значения, определяемого положением точки а. При введении третьего компонента Ж2, который является растворителем для полимера, в количестве, отвечающем точке ϐ, концентрация полимера в растворе и соответственно степень набухания его определится положением точки ϐ на соответствующей кривой равновесия.
Схематически изменение степени набухания при введении нового компонента можно передать диаграммой состояния трехкомпонентной системы (рисунок 4). Пусть чистая низкомолекулярная жидкость Ж1 вызывает набухание полимера П до значения, определяемого положением точки а. При введении третьего компонента Ж2, который является растворителем для полимера, в количестве, отвечающем точке ϐ, концентрация полимера в растворе и соответственно степень набухания его определится положением точки ϐ на соответствующей кривой равновесия.
Степень набухания аморфных полимеров обычно возрастает с температурой, что объясняется увеличением подвижности компонентов системы. Но для некоторых систем, например целлюлоза (или ее низкозамещенный эфир) — вода, степень набухания понижается с повышением температуры. Это является следствием разрушения при повышенных температурах водородных связей между водой и полимером.
Степень набухания аморфных полимеров в существенной степени зависит от предыстории образца. В пленках и волокнах, полученных испарением растворителя из однофазных растворов полимера, могут сохраниться внутренние напряжения, возникшие в момент перехода в стеклообразное состояние. При этом промежуточные значения степени набухания могут оказаться более высокими, чем равновесная величина ее (рисунок 1, кривая 2). Более значительным оказывается влияние на набухание предыстории образца полимера в тех случаях, когда при его получении система застудневала. В студне, полученном из раствора полимера в результате охлаждения или добавления вещества, не являющегося растворителем, при высушивании сохраняются большие внутренние напряжения, вызванные неравновесным характером удаления растворителя, а также распадом системы на две фазы. Набухающий образец стремится восстановить тот объем и форму, которые были присущи студню перед высушиванием. Так, степень набухания желатины тем выше, чем ниже была исходная концентрация раствора, из которого получался студень. Если же получить образец желатины из раствора путем испарения воды при температуре выше точки застудневания (расположенной в интервале 33—38 °С), то степень последующего набухания в воде резко уменьшается.
Гистерезисные эффекты при набухании полимеров, полученных высушиванием студней, отчетливо проявляются также при растворении желатины. Если пластинку желатины (или столярного клея) погрузить в горячую воду, то на ее поверхности образуется слой гомогенного концентрированного раствора, который препятствует проникновению воды в толщу пластинки, и требуется очень продолжительное время для перевода всей массы образца в раствор. С другой стороны, набухание пластинки в холодной воде протекает очень быстро благодаря релаксационному восстановлению объема, который студень занимал до сушки. Проникновение воды внутрь пластинки способствует последующему быстрому растворению желатины при нагревании.
Степень набухания трехмерных полимеров зависит от частоты сетки. Так, для слабовулканизованного каучука степень набухания в углеводородах может составлять 500— 1000%, для эбонита (высокая частота сетки) — 150— 250%. При соблюдении определенных условий по степени набухания сшитого полимера можно оценить частоту сетки.
Набухание кристаллизующихся полимеров.
Полностью закристаллизованный полимер не должен, в принципе, набухать в жидкостях. Однако вследствие того, что реально ни один из полимеров полностью не кристаллизуется, и благодаря наличию дефектов в кристаллитах кристаллические полимеры также набухают. Если по условиям получения полимера кристаллизация не была завершена до момента стеклования его аморфной составляющей, то набухание в жидкости может привести к смещению точки стеклования ниже температуры опыта и к дополнительной кристаллизации аморфной части полимера. При этом кривая в координатах степень набухания — время может иметь максимум (то есть происходит уменьшение степени набухания во времени).
Практическое значение явления набухания.
Набухание является необходимой стадией во многих процессах модификации и переработки полимеров, например, при их пластификации. При модификации полимеров в результате набухания облегчается доступ реагентов внутрь частиц полимера.
Например, в производстве вискозного волокна и целлофана для ускорения образования ксантогената целлюлозу обрабатывают водными растворами щелочей, вызывающими сильное набухание волокон. В связи с этим большое внимание уделяется условиям получения исходного материала, поскольку на степень набухания оказывает существенное влияние предыстория полимера. Технологические процессы крашения волокон, варки древесины, дубления кожи, переработки продуктов питания и многие другие также связаны с явлениями набухания.
Во многих случаях набухание полимерных материалов нежелательно. Так, набухание природных и искусственных волокон, кожи приводит к изменению размеров и формы изделий из них после смачивания. Интенсивное набухание резины в маслах ограничивает использование изделий из натурального и некоторых видов синтетического каучука в качестве амортизационных деталей в приборах и машинах.
Упаковочные материалы из целлофана при набухании в воде не только меняют размеры, но и теряют до 50—60% первоначальной механической прочности. Лакокрасочные покрытия в результате набухания легко отслаиваются от подложки. Для предупреждения этих отрицательных явлений изделия из полимеров защищают покрытиями, стойкими в агрессивной среде, либо подвергают полимер структурной или химической модификации, в частности сшиванию. Структурная модификация, приводящая к резкому уменьшению набухания в воде, происходит, например, при ориентационной вытяжке поливинилспиртовых волокон.
КаргинВ.А., Слонимский Г. Л., Краткие очерки по физико-химии полимеров, 2 изд., М., 1967;
Тагер А. А., Физико-химия полимеров, 2 изд., М., 1968;
Козлов П. В., Физико-химия эфиро-целлюлозных пленок, М., 1948, с. 286;
JoplingD. W., J. Appl. Ghem., 6, pt. 2, 79
Автор: Каргин В.А., академик АН СССР
Источник: Энциклопедия полимеров, под редакцией Каргина В.А
Дата в источнике: 1972 год